医疗器械法规注册及计算机软硬件零售领域的合规要点
随着医疗科技的飞速发展,医疗器械与信息技术的融合日益加深。医疗器械的法规注册,以及与之相关的计算机软硬件及辅助设备的零售活动,构成了一个复杂而专业的交叉领域。本文旨在解读其核心法规要求与常见问题,为相关从业者提供指引。
一、 医疗器械法规注册的核心框架
医疗器械的注册管理,核心目标是确保产品的安全、有效和质量可控。在中国,其主要依据是《医疗器械监督管理条例》。根据风险等级,医疗器械分为第一类(低风险)、第二类(中风险)和第三类(高风险),实行分类管理。
- 注册/备案流程:
- 第一类医疗器械:实行产品备案管理,向所在地设区的市级药品监督管理部门提交备案资料。
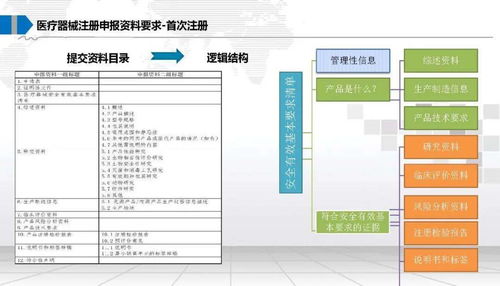
- 第二类、第三类医疗器械:实行产品注册管理。申请人需向国家药品监督管理局(NMPA)或指定的省级药品监督管理部门提交包含产品研制报告、临床评价资料、产品技术要求、风险分析报告等在内的全套注册申请资料,经技术审评、行政审批通过后获得医疗器械注册证。
- 临床评价要求:高风险产品通常需要进行临床试验,以验证其安全有效性。部分低中风险产品可通过同品种比对等方式进行评价。
- 质量管理体系:医疗器械注册申请人/注册人必须建立符合《医疗器械生产质量管理规范》的质量管理体系,并在注册过程中接受现场核查。
二、 与计算机软硬件零售的交叉与合规要点
当医疗器械的功能实现高度依赖于特定的计算机、服务器、专用硬件或软件时,这些软硬件设备便不再是普通的电子产品,其流通和使用需考虑医疗器械法规的延伸监管。
- 作为医疗器械组成部分的软硬件:
- 如果计算机或特定硬件是医疗器械整机不可分割的一部分(如内置在医疗设备中的控制计算机、图像处理工作站),其安全性、有效性已包含在整机的医疗器械注册证中。零售商销售此类“整机”时,必须确认其已取得有效的医疗器械注册证。
- 如果软件本身被界定为独立软件医疗器械(如影像处理软件、辅助诊断软件),则无论以何种载体(光盘、U盘、在线下载)提供,该软件本身必须完成医疗器械注册或备案。零售商销售此类软件,实质上是在销售医疗器械。
- 作为医疗器械辅助设备的软硬件:
- 许多通用计算机、服务器、显示器、网络设备等,被用作已注册医疗器械的显示、存储、传输终端。此类零售活动属于普通的“计算机软硬件及辅助设备零售”。
- 核心合规点在于:零售商需明确告知客户,这些通用产品并非医疗器械,不能单独用于医疗目的,其性能和配置需满足其所连接医疗器械的制造商明确规定的技术要求。避免宣传其具有医疗功能,以防构成未经注册的医疗器械的虚假宣传或非法销售。
- 数据安全与隐私保护:用于处理医疗数据的计算机和存储设备,需符合《网络安全法》、《数据安全法》及《个人信息保护法》的要求,确保医疗健康数据的安全存储与传输。零售商在提供解决方案时,应提示客户相关法律义务。
三、 常见问题与风险提示
1. 问题:销售用于医疗环境的通用电脑,需要医疗器械经营许可证吗?
解读:如果该电脑本身不具备独立的医疗功能,未被定义为医疗器械,仅作为通用计算平台,则其销售不需要医疗器械经营资质。但应确保其质量可靠,并建议客户遵循医疗器械制造商对配套设备的技术规范。
2. 问题:为医院信息系统(HIS)提供服务器集成服务,涉及法规吗?
解读:需区分情况。如果HIS系统本身是取得注册证的医疗器械软件,那么为其提供定制化集成服务可能被视为医疗器械经营活动的一部分,需与系统注册人明确责任划分。如果仅为通用IT基础设施部署,则不直接受医疗器械法规管辖,但需严格遵守医疗行业的数据安全和等保要求。
- 风险提示:最大的法律风险在于“模糊地带”的违规。
- 非法销售未经注册的医疗器械:将具有医疗功能(如诊断、治疗、监测)的软件或软硬件一体设备,作为普通商品销售。
- 虚假宣传:对通用的计算机硬件进行具有医疗效果的宣传,误导消费者。
- 质量责任:提供的辅助设备因质量问题导致连接的医疗器械数据错误或功能失效,可能需承担相应的民事及产品质量责任。
四、 与建议
在医疗器械与信息技术深度融合的背景下,从事相关软硬件零售和服务的企业,必须建立清晰的合规边界意识:
- 准确分类:严格区分所售产品是“医疗器械”本身,还是“医疗器械的通用辅助工具”。
- 资质查验:销售医疗器械(包括独立软件),必须查验并留存有效的医疗器械注册证和供应商的医疗器械经营许可证。
- 宣传合规:宣传内容实事求是,不对非医疗器械产品明示或暗示医疗功能。
- 强化合同与告知:在销售合同中明确产品属性、用途限制,并向客户提供必要的技术符合性说明与风险提示。
- 关注数据安全:主动了解和满足医疗行业对信息安全的特殊要求。
通过建立完善的内部合规流程,相关企业不仅能有效规避法律风险,更能赢得医疗机构和患者的信任,在医疗健康产业数字化浪潮中行稳致远。
如若转载,请注明出处:http://www.dhguiks.com/product/66.html
更新时间:2026-06-19 02:41:26